Installation
To use STEGO, first install R, RStudio and other required programs
Please install R version 4.3.2 or greater. If your operating system doesn’t support this version of R, than some of the functions (e.g., annotation with scGate) may not work as expected.
MAC:
Windows:
Linux:
install R.linux
using sudo apt install the following: libcurl, libfontconfig1, libtiff5, liblapack-dev, libblas-dev, libglpk-dev, libudunits2-dev, libgdal-dev libgeos-dev, libproj-dev and also the gfortran
Sytem requirements
Recommended to use a 64-bit OS with at least 16GB of RAM, ideally >=32GB. This is needed for the memory intensive merging and annotation steps.
When running STEGO.R merging and annotation steps, we recommend to limit the number of other running programs to help prevent RAM memory overflow, espeically if you have <32Gb of RAM.
Storage requirements will depend on the number of cells annotating. For example, at least 200 GB of free space on a Macbook Pro was needed to process the T cell Atlas (V2/V3).
Download the project directory
From the github repository STEGO.R
Download the Directory_for_Project.zip
Unzip the Directory_for_Project
Rename the folder, if desired.
This folder contains instructions on how to process the data and will be used to set up the R.project environment.
In the Directory_for_Project open up the Project.Rproj i. Quick tutorial for understanding RStudio environment with this YouTube video.
short-cut key for running a line of R code
put the cursor on the line you want to run.
Mac: Command shift Enter
Windows: ctrl shift Enter
Downloading and installing STEGO.R
Follow the steps in the install.R file found in the Directory_for_Project
# see https://stegor-documents.readthedocs.io/en/latest/index.html for more details on the tutorial to follow through.
# This is to make sure with installing from source is set to the g++ compiler
Sys.setenv("CXX" = "g++")
# installing STEGO.R
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("devtools", quietly = TRUE))
install.packages("devtools")
if (!require("usethis", quietly = TRUE))
install.packages("usethis")
# install STEGO.R with Devtools
# if you get asked about installing from SOURCE, select NO.
# if asked about updating the packages, select either 1 for all, or 3 for none.
# also run this line to check for updates
devtools::install_github("KerryAM-R/STEGO.R")
# if asked to update the app either select 1 to updated all
1
# if asked to install from source on the M1 Mac chip (Seurat)
y
# if then prompted with all the bioconductor based packages to install from source on M1 or greater Mac chip
y
# under development - latest build, but may be unstable
# devtools::install_github("KerryAM-R/STEGO.R", ref = "beta-version", force = T)
# import all of the fonts to your system. Will only need to be done the first time.
require(extrafont)
font_import()
# when it asks if you would like to install the different fonts
y
loadfonts()
fonts <- fonttable()
head(fonts)
# you many need to add in the git_token, and the Renviorn is opened with the following code.
usethis::edit_r_environ()
STEGO.R not found/API issue
You will need to create a git token
# Create the token
usethis::create_github_token()
# Open the .Renviron file
usethis::edit_r_environ()
# Paste your token into the file
GITHUB_PAT=insert_your_token
# Close the file and restart R
Possible installing prompts
When you see this line: "These packages have more recent versions available. It is recommended to update all of them. Which would you like to update?"
Answer with "1" and hit "Enter".
Other Mac prompts on M1 or greater chip
The installation may ask you the following to install from source. In this instance, select yes.
There is a binary version available but the source version is later:
binary source needs_compilation
Seurat 5.0.3 5.1.0 TRUE
Do you want to install from sources the package which needs compilation? (Yes/no/cancel)
y
Installing the bioconductor packages from Source.
Packages which are only available in source form, and may need compilation of C/C++/Fortran: ‘zlibbioc’ ‘XVector’ ‘SparseArray’ ‘S4Arrays’ ‘IRanges’ ‘Biobase’ ‘DelayedArray’ ‘GenomicRanges’ ‘S4Vectors’
‘BiocNeighbors’ ‘BiocParallel’
Do you want to attempt to install these from sources? (Yes/no/cancel)
y
Expected final lines once STEGO.R has been correctly installed.
R CMD build
✔ checking for file ‘/Users/kerrymullan/Desktop/STEGO_copy.R/Temp/Rtmp0n6xNi/remotes176117593b632/KerryAM-R-STEGO.R-df4640eae1a05f44f3c91ca527215f4af56894ff/DESCRIPTION’ ...
─ preparing ‘STEGO.R’:
✔ checking DESCRIPTION meta-information ...
─ checking for LF line-endings in source and make files and shell scripts
─ checking for empty or unneeded directories
─ building ‘STEGO.R_1.0.0.tar.gz’
* installing *source* package STEGO.R’ ...
** using staged installation
** R
** inst
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
*** copying figures
** building package indices
** testing if installed package can be loaded from temporary location
** testing if installed package can be loaded from final location
** testing if installed package keeps a record of temporary installation path
* DONE (STEGO.R)
.rs.restartR() # restart R
Installing fonts
To have access to wider variety of font, you will need to install the following found in the Install.R folder
# import all of the fonts to your system. Will only need to be done the first time.
require(extrafont)
font_import()
# when it asks if you would like to install the different fonts
y
loadfonts()
fonts <- fonttable()
head(fonts)
Running STEGO.R
Now the the R environment is set up and the STEGO.R and it’s dependencies are installed, we can now run the application.
Run the following lines in R. A window will opened that runs the STEGO.R shiny R application.
require(STEGO.R)
runSTEGO()
You are now ready to process your scRNA-seq with scTCR-seq data!
Note, if a window doesnt open, you will need to troubleshoot which packages haven’t been installed.
Trouble shooting missing packages
Check that you have the latest version of R.
Check that you downloaded first the devtools and bioconductor.
API issue with creating a github token.
Understanding the “Directory_for_Project” Folder
Unzip the “Directory_for_Project.zip” file. This folder contains our recommended structure on how to organise your files for each scRNA-seq project. We will refer to these files through out the Tutorial.
This folder contains the following folder:
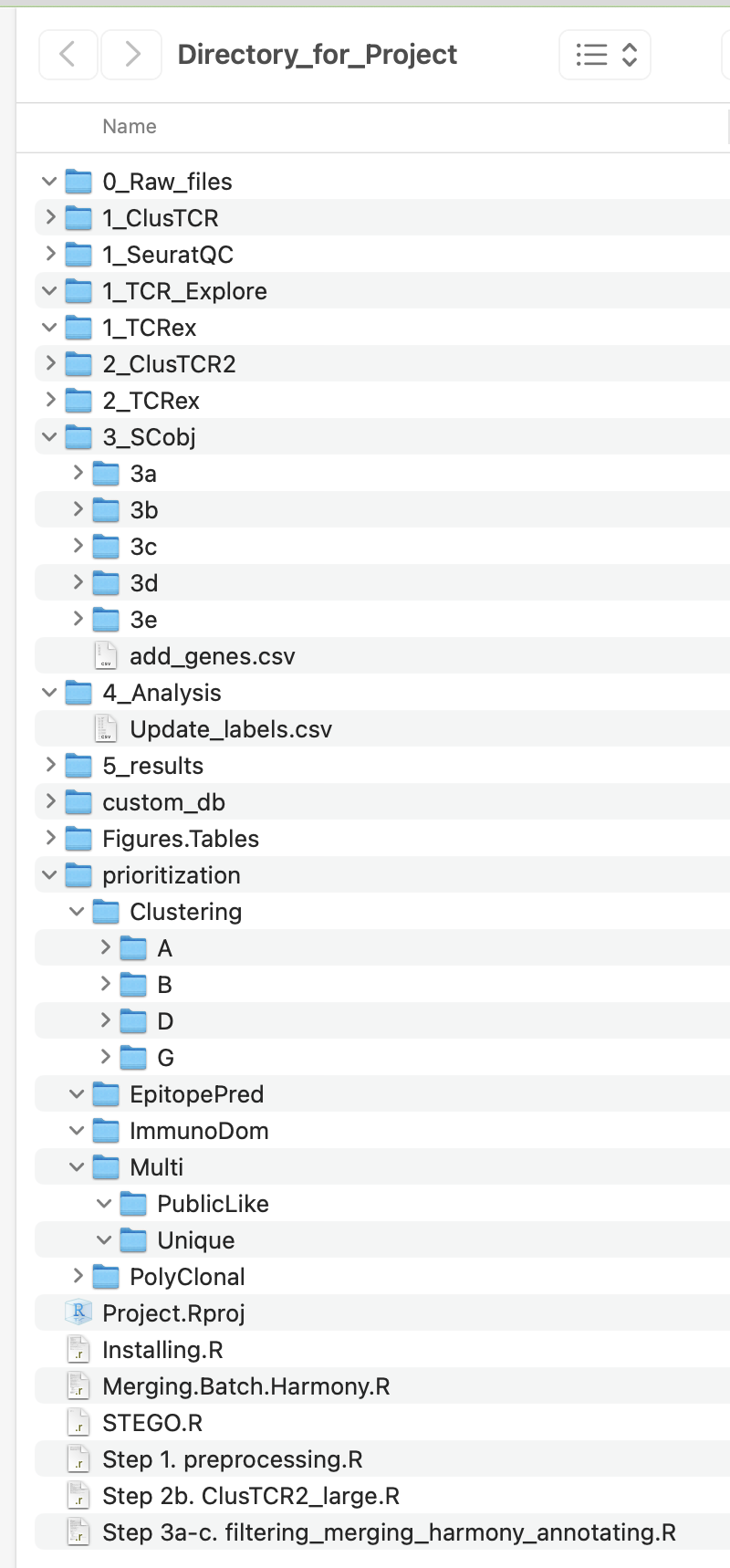
The user will also have access to the following files:
Installing.R
1_preprocessing.R (10x only)
2b_ClusTCR2_large.R
3a-c_filtering_merging_harmony_annotating.R
Project.Rproj
STEGO.R
Re-installing
Re-run this line from the install.R file to check for updates.
Restart R, once it has been installed. If you have the latest version of R, updated all packages with 1. However, if R is out of date, use 3 instead.
devtools::install_github("KerryAM-R/STEGO.R")