Step 2. TCRex and ClusTCR2
Note
- Consideration for the analysis.
The pipeline was built for scRNA-seq with scTCR-seq datasets.
QC documentation is being actively updated.
- Prerequisite
Install STEGO.
Copy all your raw files to the 0_Raw_files folder within your project.
Downloaded files to 1_ClusTCR2, 1_SeuratQC, 1_TCRex and TCR_Explore
2a. Merging TCRex
TCRex is a user-friendly webtool designed to facilitate the prediction of epitope-TCR binding. It starts from a TCR file containing a list of TCR beta sequences. After selecting the epitopes of interest, TCRex predicts the binding between every TCR sequence and every epitope. Currently it supports predictions for 100 epitopes. More information on how to use this tool and interpret the results can be found at TCRex website. 1. Upload all the files stored in the "1_TCRex" for merging and save the merged file to the 1_TCRex folder. 2. The TCRex merge file can then be uploaded to the TCRex website to predict if the beta chain will interact with certain epitopes. 3. Once the TCRex process is complete, download the processed file, and move it to the 3_Analysis folder.
2b. Clustering with ClusTCR2
ClusTCR2 is an R alternative to the python package ClusTCR [1], specifically developed to be applied to single cell TCR sequencing data. The original version of ClusTCR applies a two-step process to cluster large sets of TCR sequences. The first step involves encoding TCRs into a numerical vector, based on the physicochemical properties of the amino acids. K-means clustering is applied to the amino acid vectors to create large families of pre-clusters. During the second step, a hashing function is used to identify all pairs of CDR3 amino acid sequences with ≤ 1 Hamming distance mismatch. From these CDR3 pairs, a graph is constructed, and communities are detected using the Markov clustering (MCL) algorithm, which will be the final clusters.
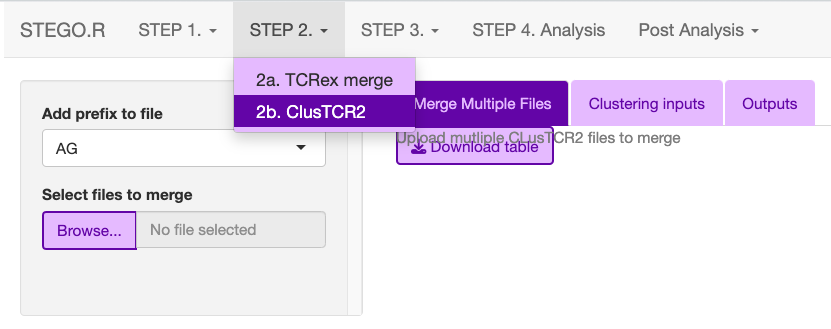
This process requires the user to upload AG separate for the BD clustering. This is due to how the data is re-combined with the meta data in the Seurat object in STEP4. Analysis.
- If there are multiple files under the ‘ClusTCR2’, merge the AG_ or BD_ from the 1_ClusTCR2 folder.
save the file with AG_ or BD_ prefix.
Upload either the merged file or single AG_ or BD_ under the “Clustering inputs” tab.
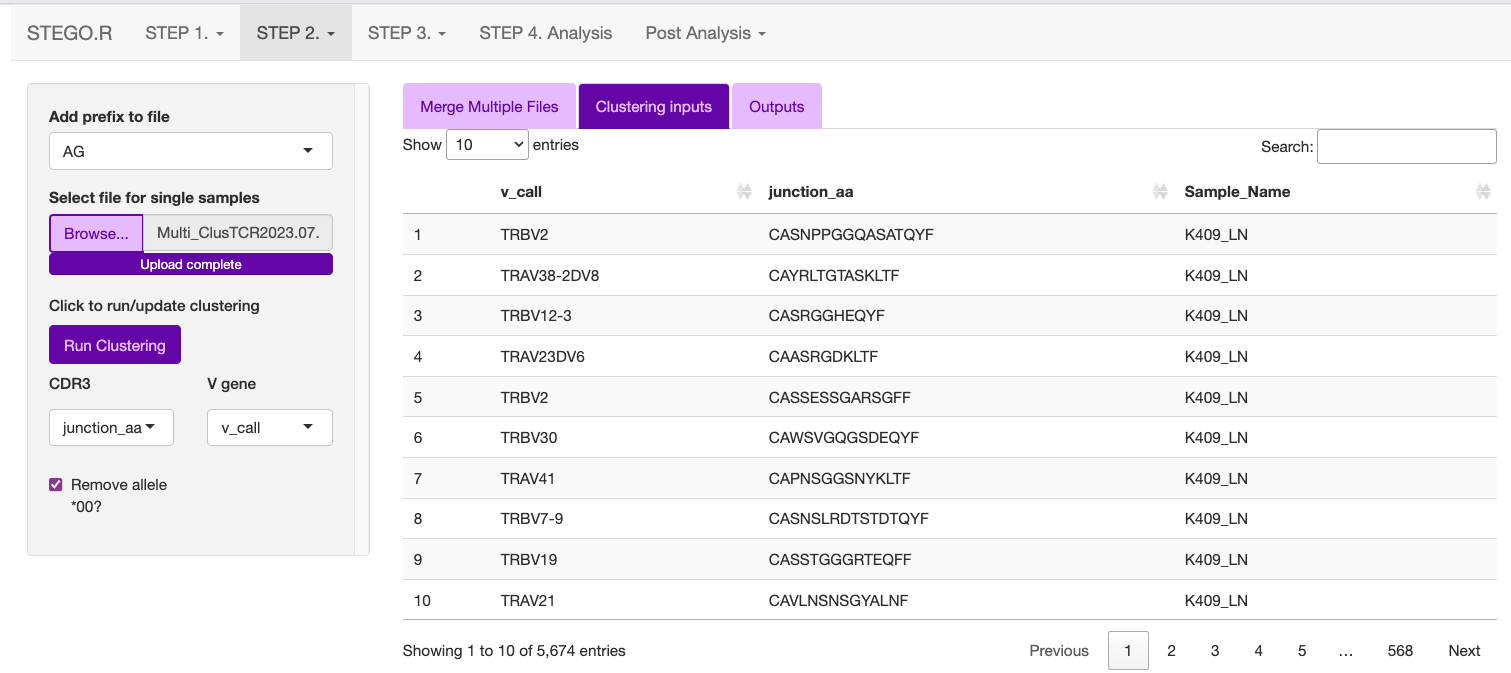
Hit the “Update clustering” button to perform the clustering.
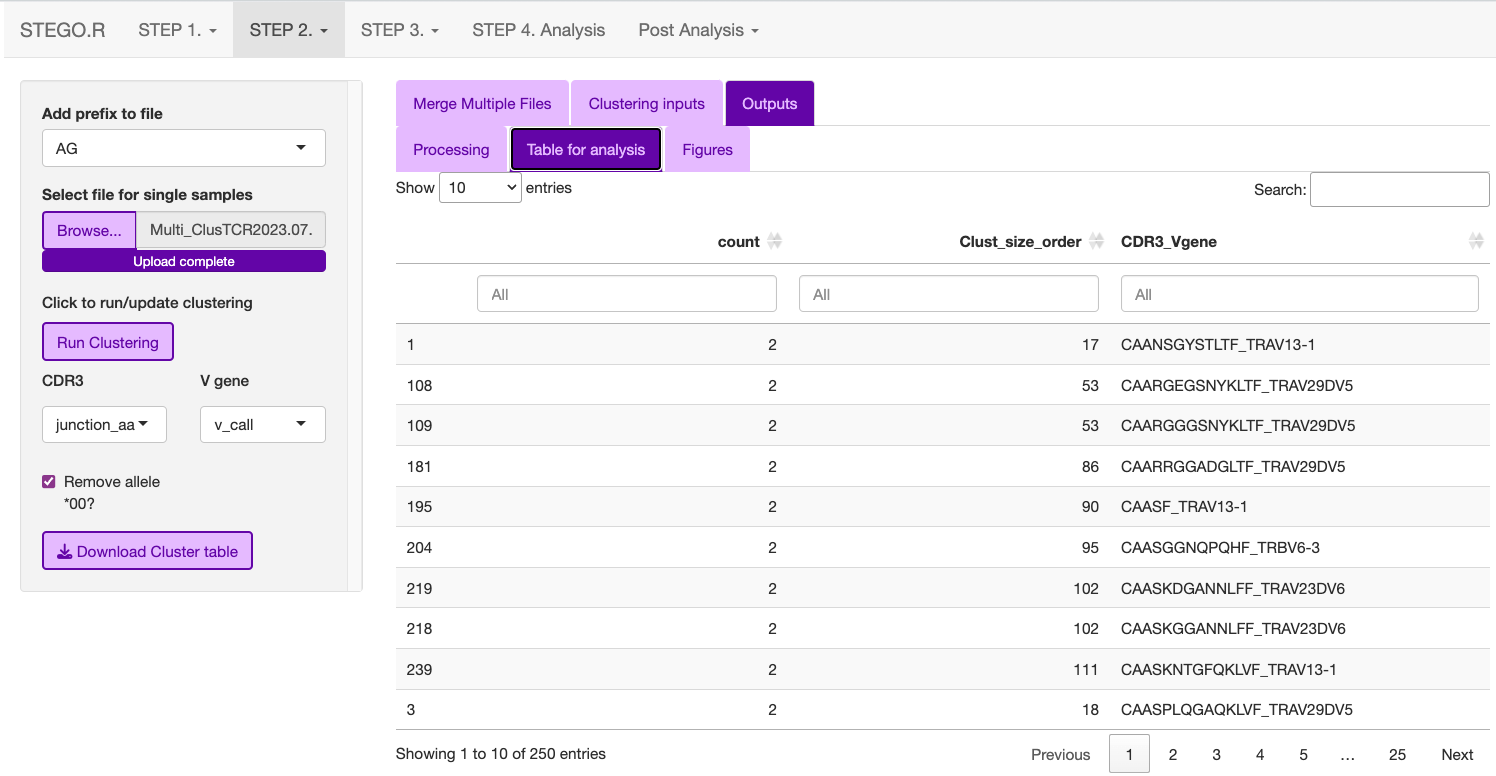
- Under the “output” tab has three sub-sections
Processing: Time taken to complete the clustering
Table for analysis: Download the table to the “3_Analysis” section; make sure you add the AG_ or BD_ prefix to the file name.
Figures: The user can explore the data and download the clustering network and the corresponding motif. The latter figure is also generated in the “Analysis section”.
Command line process
Step 2b. has command line equivalent codes.
require(ClusTCR2)
# this process assumes that you have already merged the files using the interface, and saved the file in 1_ClusTCR folder
# alpha/gamma chain -----
clust.data.raw <- read.csv("1_ClusTCR/AG_Multi_ClusTCR.csv")
# this should print the v_gene name
names(clust.data.raw)
# once you have checked the v_gene name matches, proceeded to runing the clustering step (1 edit distance), followed by the mcl step to label the clusters.
step1 <- ClusTCR2::ClusTCR_Large(clust.data.raw,allele = F,v_gene = "v_gene")
step2 <- ClusTCR2::mcl_cluster_large(step1)
# this saves both the analysis cluster table and the list object that can create each of the unique network plots
saveRDS(step2,"1_ClusTCR/AG_clusTCR2.all.rds")
# saves the final clustering table for the Step 4. Analysis section.
write.csv(step2[[1]],"AG_clusTCR2.csv",row.names = F)
# beta/delta chain ----
clust.data.raw <- read.csv("1_ClusTCR/BD_Multi_ClusTCR.csv")
names(clust.data.raw)[2]
step1 <- ClusTCR2::ClusTCR_Large(clust.data.raw, allele = F, v_gene = "v_gene")
step2 <- ClusTCR2::mcl_cluster_large(step1)
saveRDS(step2,"1_ClusTCR/BD_clusTCR2.all.rds") # saves the two list objects
write.csv(step2[[1]],"3_Analysis/BD_clusTCR2.csv",row.names = F)
References 1. Valkiers, S. et al. ClusTCR: a python interface for rapid clustering of large sets of CDR3 sequences with unknown antigen specificity. Bioinformatics, 2021. 37(24): p. 4865-4867. 2. Gielis, S. et al. Detection of Enriched T Cell Epitope Specificity in Full T Cell Receptor Sequence Repertoires. Front Immunol 10, 2820 (2019).